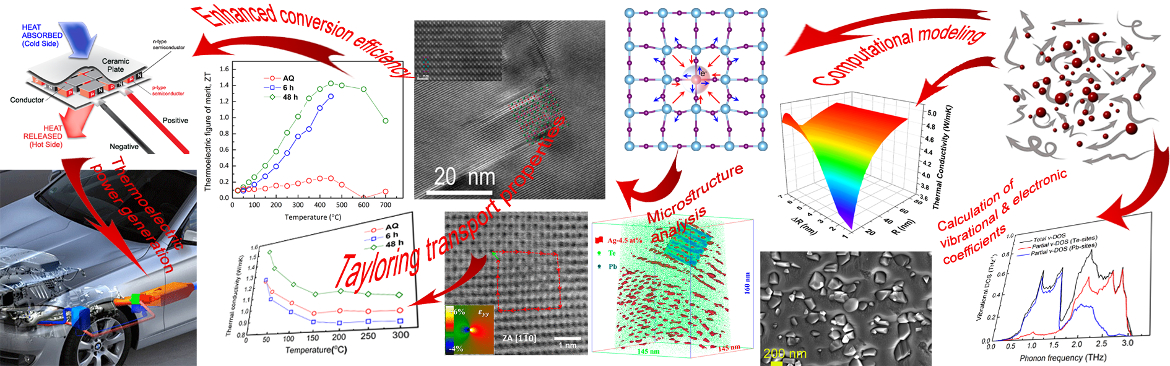
Microstructure, chemistry, and transport of charge and heat: a story of materials design
Understanding the underlying physics of thermoelectric materials
One of the fundamental and intriguing questions in materials science is how to control functional properties of materials by manipulating their microstructure. An outstanding example for a functional property that is highly sensitive to the finest features of microstructure is thermoelectricity. In the thermoelectric (TE) effect thermal energy is converted into electrical energy and vice-versa. TE devices can, therefore, serve for heat-exchange or refrigerating as well as for capturing waste heat and converting it into electricity; the latter has major implications for energy harvesting.
Development of TE materials with enhanced power generation capabilities begins with controlled doping of semiconductors to optimize the Seebeck coefficient (thermopower) and electrical conductivity; both inversely depend on the doping level. Further improvement of the TE performance is achieved by increasing their electrical conductivity and/or reducing their thermal conductivity. These two requirements are usually contradictory, which makes the development of TE materials a grand challenge from the materials science point of view. One of the promising approaches for reducing thermal conductivity with no deterioration of the electrical conductivity is formation of coherent nanometer- size precipitates dispersed in a TE matrix, thereby scattering of heat conducting phonons. Today, TE materials research introduces new questions related to materials synthesis, thermodynamic stability of materials, design of micro-to-nanometer size features in the bulk, materials characterization, microstructure evolution, thin film and micron-size devices, quantum confinement effects in nanoscopic materials, energy filtering of charge carriers, and more.
We are interested in understanding the underlying physics of how materials’ microstructure and chemistry determine functional properties, and in the establishment of basic rules for materials design. We process single-phase or two-phase TE compounds comprising matrix and precipitates, and apply different heat treatments to obtain diverse microstructures. Supplemented by ab-initio calculations applying the density functional theory (DFT), we aim to “smartly-design” new materials a-priori in a time and cost-effective manner. Employing advanced characterization techniques, we analyze the materials structure and chemistry down to the nanometer length-scale. Finally, we measure the TE properties of these materials and correlate them with their microstructures. This helps us modeling the thermal conductivity in heterogeneous materials, and provides us with insight of the underlying physics. Besides possible improvements of TE properties, we endeavor to advance our understanding of how functional properties of materials depend on their microstructure.

The Thermoelectric Materials Group: Research Directions
We investigate diverse types of TE bulk materials, which can be coarsely classified into telluride- and oxide-based materials. Whereas the first group comprises materials featuring the highest values of TE conversion efficiency, they are toxic, volatile, and expensive. However, these materials offer the best solution for both cooling and power generation up to mid-range temperatures. Conversely, oxides are abundant, environmentally-friendly, chemically stable at elevated temperatures, and inexpensive. The downside is that they are significantly inferior to tellurides in terms of TE efficiency. Remarkably, oxides are the best solutions for power generation at elevated temperatures up to 1000 °C.
Nanostructured two-phase lead telluride (PbTe) based compounds: We alloy PbTe with Ag at different concentrations to enable controlled nucleation of the Ag2Te-rich phase via aging heat treatments, resulting in Ag2Te-precipitates of different size and number density dispersed homogeneously in the PbTe-based matrix. The immediate objective of such microstructures is scattering of phonons; however, the side-effects of precipitate growth or dissolution processes are either consumption of Ag-atoms from the matrix or enrichment with Ag-atoms, respectively. This, of course, affects the electron transport properties, since Ag is not only a second-phase forming agent, but also acts as a dopant. Applying transmission electron microscopy (TEM) in combination with atom probe tomography (APT), we could observe the entire sequence of nucleation, constant-rate growth, and coarsening of Ag2Te-based precipitates as well as other Ag-rich phases. This combination of techniques allowed us to observe precipitate number density values as high as ca. 1024 m3 together with the quantification of the Ag-concentration inside the inter-precipitate regions in the < 0.1 at. % range, thereby providing a reasonable elucidation of the behavior of the full set of TE transport coefficients and ways to control them upon processing conditions and heat treatments, while addressing thermal stability questions. Ag atoms decorating dislocation cores also affect TE transport. Ostwald ripening of Ag2Te-precipitates is determined by the PbTe/Ag2Te interface crystallography in terms of orientation relationships. APT also enables us analyzing the probability of multiple evaporation events, which is indicative of the nature of inter-atomic bonding, revealing that the PbTe-matrix is characterized by meta-valent bonding, whereas the Ag2Te phase comprises iono-covalent bonding; this contrast gives rise to more efficient phonon scattering at PbTe/Ag2Te interfaces. Further details: Sheskin et al. ACS Appl. Mater. Inter. 2018 ; Yu et al. ACS Appl. Mater. Inter. 2018 ; Amouyal et al. Theory Simul. 2018; Dahan et al. Acta Mater. 2020 ; Yu et al. 2024.

Interface engineering of bismuth telluride (Bi2Te3) based compounds: We apply the topologically insulating (TI) nature of Bi2Te3 to control its TE transport by interface engineering. Topological surface states possess remarkably high electron mobility due to protection by time-reversal symmetry, rendering their charge transport inert against impurity, oxidation, or any undesirable phenomena (in the absence of magnetic field). We observe TI behavior in Bi2Te3 produced by compaction and hot-pressing of nanoparticles at different conditions, resulting in polycrystalline Bi2Te3 of different sub-micron grain sizes. Under certain conditions, electron mobility increases with decreasing grain size, which is counter-intuitive and is associated by us with topological surface states. Doping with 3 at. % Nd eliminates the TI nature, possibly due to its magnetic moment. We also investigate the effects of crystallographic grain boundary (GB) misorientation on TE and TI behaviors, showing that GBs of special misorientations exhibit TI effect. We optimize materials synthesis by producing bulk Bi2Te3 from molecular precursors in liquid solutions, e.g. synthesis from Bi2O3 + Na2TeO3 precursors results in n-type Bi2Te3 containing a high number density (Nv ∼45 × 1023 m−3) of elemental Te-nanoprecipitates decorating the Bi2Te3 GBs, which yields enhanced TE power factor of ∼19 μW cm−1 K−2 at 300 K and a promising TE figure of merit (zT) peak value of 1.30 at 450 K. Further details: Baranovskiy et al. Adv. Theory Simul. 2019 ; Solomon et al. ACS Appl. Nano Mater. 2021 ; Gayner et al. ACS Appl. Mater. Inter. 2022 #1 ; Gayner et al. ACS Appl. Mater. Inter. 2023.

Design of CaO(CaMnO3)m– based thermoelectric oxides: CaO(CaMnO3)m– based compounds belong to the Ruddlesden-Popper (RP) group and feature special crystal structures with repeating m– perovskite CaMnO3-units separated by single rock-salt CaO-layers. For example, an ‘m=2’ structure contains the following repeating units: …-(CaO)-(CaMnO3)-(CaMnO3)-(CaO)-(CaMnO3)-(CaMnO3)-(CaO)… In this terminology, m=∞ corresponds to the absence of such interfaces (planar density of the CaO-layers equals zero), and m=1 represents the maximum possible planar density. We demonstrate both experimentally and computationally that varying the m-values is accompanied by marked effects on both phonon and electron transport, so that increasing the density of the CaO- atomic planes (reducing m) reduces both thermal and electrical conductivities due to phonon and electron scattering, respectively, and vice-versa. We further focus on the role of interphase boundaries in CaMnO3/Ca2MnO4 composites and on atomistic mechanisms of heat and charge transport in these compounds, attempting to derive universal rules. We investigate the vibrational properties of these compounds, demonstrating that, besides the direct effects of CaO-planes on phonon scattering, acoustic phonon modes in the entire CaO(CaMnO3)m series are dominated by Ca-atom oscillations. For this reason, doping at Ca-sublattice sites scatter phonons more effectively than doping at Mn-sites; also, inelastic Umklapp scattering is governed by the degree of anisotropy of both Grüneisen parameters and sound velocities. On the electronic aspect, we hypothesize that the CaO-planes act as energy barriers for electron transport. We, accordingly, apply selective doping to reduce barrier heights, thereby facilitating thermal activation of charge carriers and enhancing their mobility. On the broader view, we seek correlations between electron transport in these oxides and their vibrational and elastic properties, hypothesizing that the former is governed by polaron hopping. Employing the small polaron hopping model for Ca2MnO4 (m=1), we found that the polaron hopping energies of Y-doped compounds are lower than their La-doped counterparts, and attain minimum values of 60 and 73 meV, respectively. This is associated to softening of interatomic bonds, which is more pronounced for Y-doping compared to La-doping. Our studies demonstrate that charge carrier dynamics are strongly coupled with elastic properties, implying that the latter can be used to predict charge carrier mobility in oxide semiconductors. We observe similar correlations between lattice softening and enhanced charge transport in the compounds CaMnO3 (m=∞) and Ca3Mn2O7 (m=2), which enables us to generalize our conclusions. Further details: Baranovskiy et al. Nano Energy 2018 ; Azulay et al. Acta Mater. 2019 ; Azulay et al. ACS Appl. Mater. Inter. 2020 ; Natanzon et al. Israel J. Chem. 2020 ; Azulay et al. Acta Mater. 2023 ; Azulay et al. Amer. Ceram. Soc. 2023 ; Azulay et al. Adv. Energy Sust. Res. 2024
Design of multiphase ZnO-based thermoelectric oxides: We study single- and multi-phase ZnO based compounds, which exhibit the best TE performance among oxides. Herein, we apply the energy filtering (EF) effect, in which low-energy charge carriers are selectively filtered-out due to inclusions or second-phase precipitates dispersed in the TE matrix. We, accordingly, design quaternary oxides based on ZnO co-doped with Al and Ti, forming a three-phase ZnO/ZnAl2O4/Zn2TiO4 We observe unusual behavior, where both Seebeck coefficient and electrical conductivity values increase simultaneously with temperature due to EF effects associated to both co-doping and the peculiar multiphase structure, besides reduction of the thermal conductivity. Further details: Koresh & Amouyal. J. Eur. Ceram. Soc. 2017 ; Gayner & Amouyal. Adv. Funct. Mater. 2020 ; Gayner et al. ACS Appl. Mater. Inter. 2022 #2
