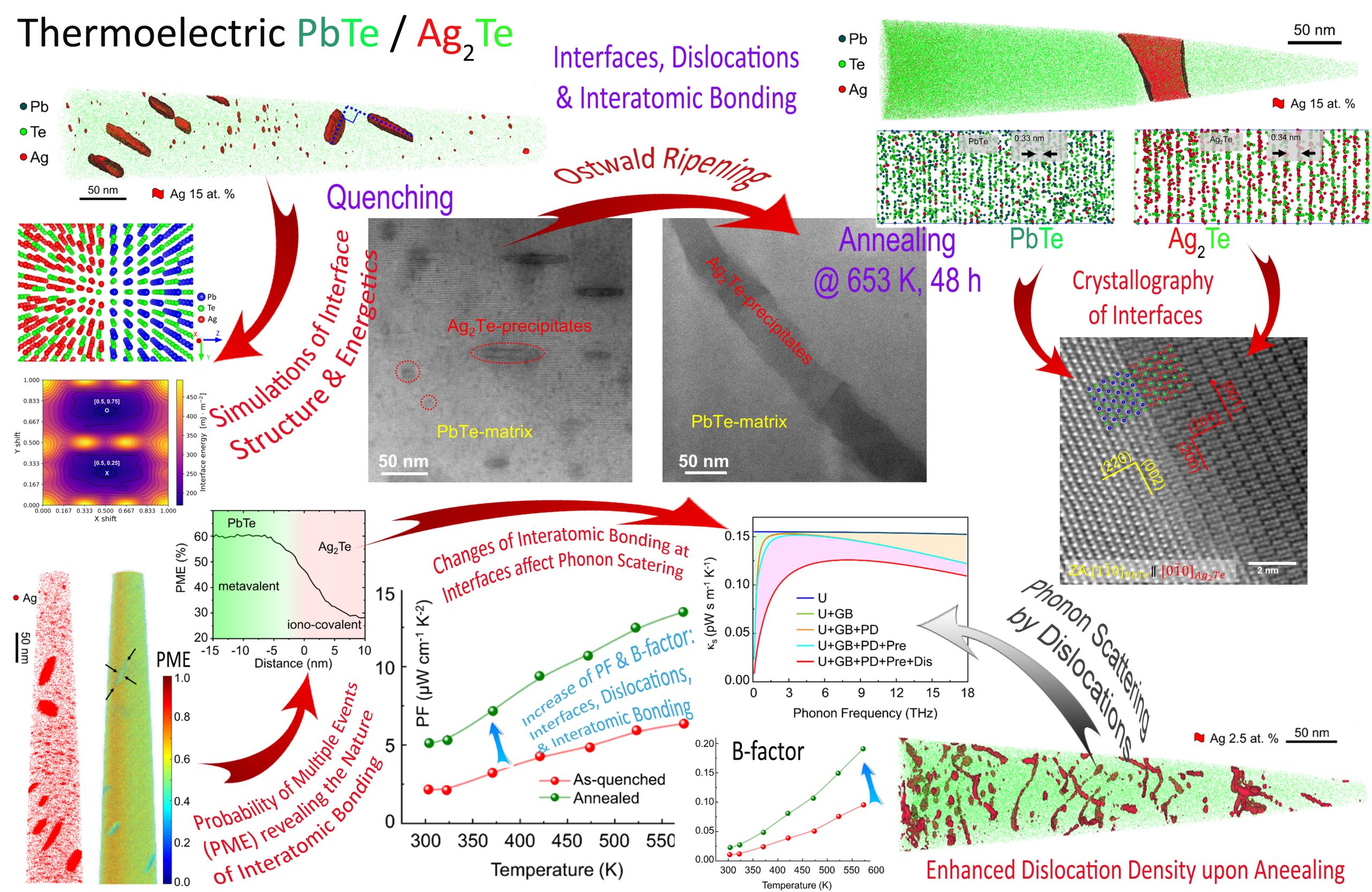
Ostwald ripening of Ag2Te precipitates dispersed in PbTe-matrix due to annealing is dictated by the atomistic structure and orientation relationship between the PbTe and Ag2Te phases, and results in enhanced dislocation density, thereby affecting thermoelectric transport. Interfacial energies of the PbTe/Ag2Te interfaces are simulated and the nature of interatomic bonding across these interfaces is evaluated based on the probability of multiple evaporation events from atom probe tomography (APT), revealing transition from metavalent to iono-covalent bonding, respectively, Adv. Energy Mater(2024).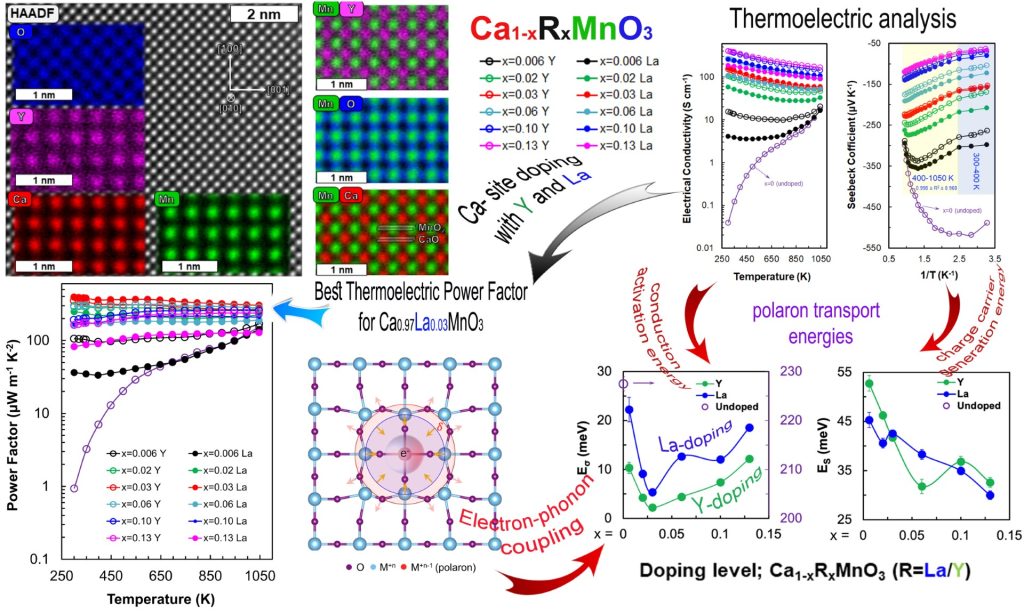
Enhancing electronic transport properties of thermoelectric oxides is of great technological importance. Oxides are promising candidates for waste heat harvesting at elevated temperatures as well as for electricity generation in low-power applications. To this purpose, fundamental understanding of their electrical and thermal conduction mechanisms is essential. Herein, the conduction mechanism of CaMnO3 materials is focused on and how dopant identity and amount alter the kinetic properties of charge transport is investigated. Ca1−xRxMnO3 compounds with R = Y and La are synthesized, where 0 ≤ x ≤ 0.13, and the electrical conductivity and Seebeck coefficient for temperatures ranging from 300 to 1050 K, indicating that Y-doped compounds are usually more conductive than their La-doped counterparts, are measured. Analysis of both in terms of the small polaron hopping model reveals that Y doping reduces conduction activation energies, resulting in higher electrical conductivity and charge carrier mobility. Remarkably high values of thermoelectric power factor for the Ca0.97La0.03MnO3 compound, for example, 300 μWm−1 K−2 at 1050 K are observed; furthermore, these values are preserved for a wide temperature range, rendering this compound a good candidate for heat-to-electrical power generation at elevated temperatures. Adv. Energy Sustainability Res(2024)
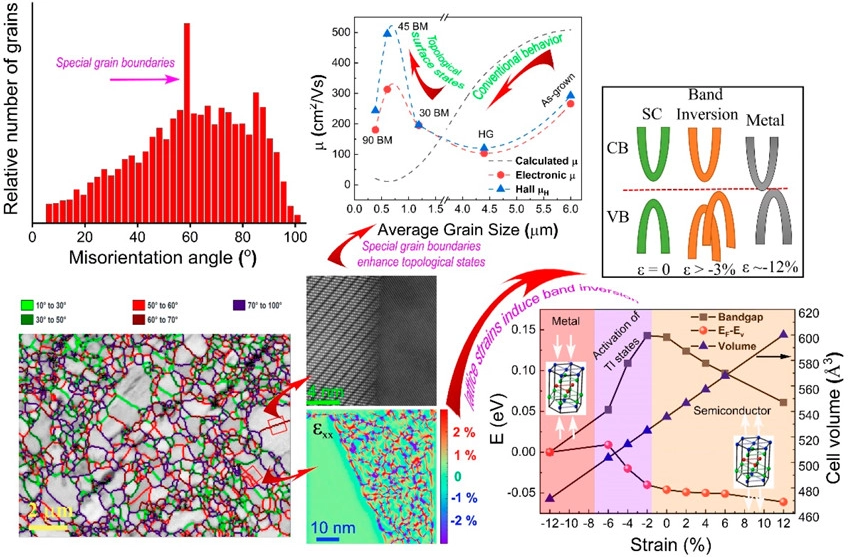
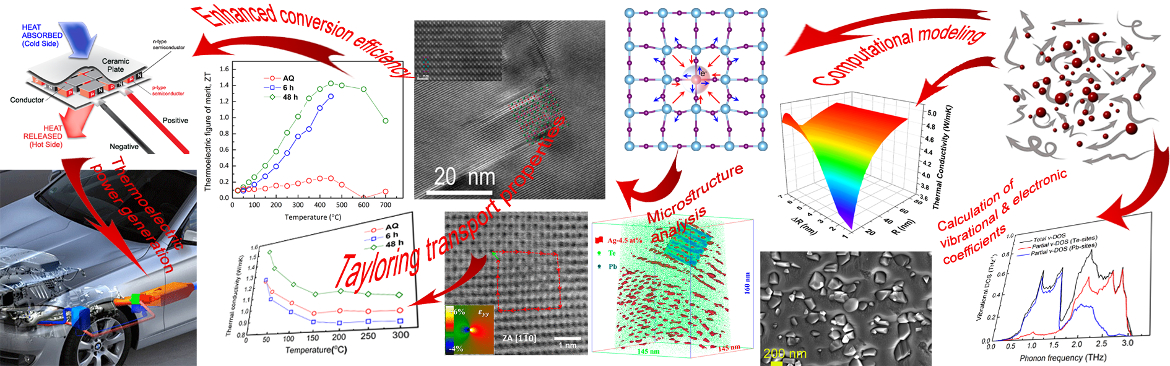
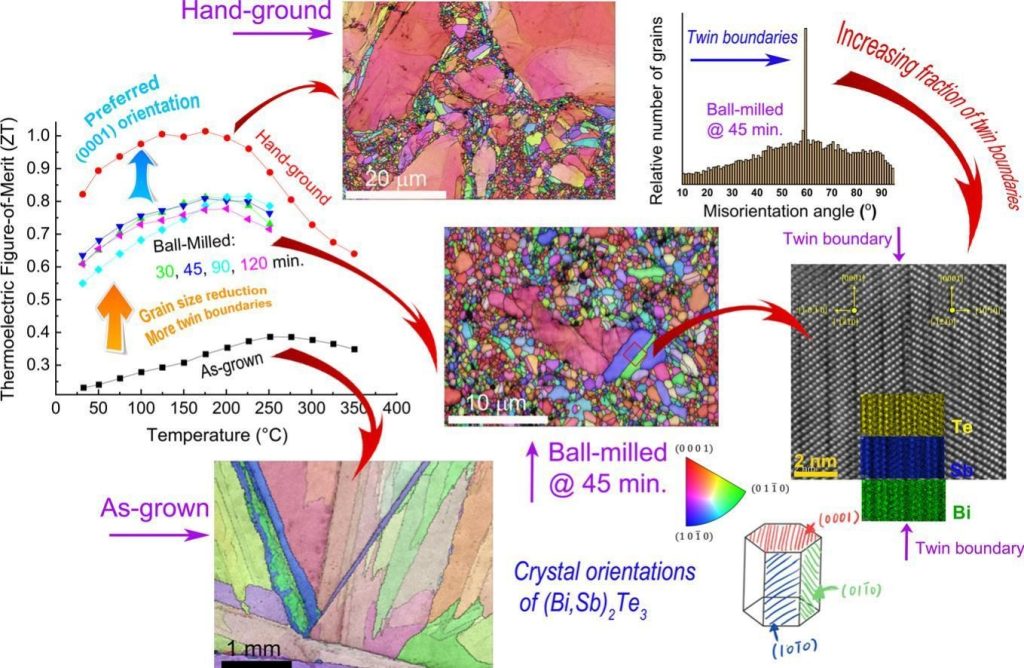
Topological insulators (TIs) and thermoelectric (TE) materials seem to belong to distinct physical realms; however, in practice, they both share common characteristics. Introducing concepts from TIs into TE materials to enhance their performance and achieve better understanding of electronic transport requires extensive research. Particularly, grain size, misorientation, and grain boundary (GB) character are of utmost importance to attain effective charge carrier transport in TE polycrystals; these factors, however, have not been thoroughly explored. Herein, we investigate the correlation between grain size, misorientation, and lattice strain in Bi2Te3 and its TI signature, aiming to improve its TE performance. We reveal an unusual behavior showing that electron mobility increases upon the increase of grain size, reaching at a maximum value of 495 cm2/V·s for an optimum grain size of 600 nm and most-frequent GB misorientation angle of 60° and then decreases with increasing grain size. It is also indicated that the combined effects of grain size reduction and point defects induce lattice strain in the Bi2Te3-matrix that is essential to trigger the TI contribution to TE transport. This trend is corroborated by first-principles calculations showing that compressive strains form multiple valleys in the valence band and opens the TI band gap. Such a combination of physical phenomena in a well-known TE material is unique and can promote our understanding of the nature of TE transport with implications for TE energy conversion. C.Gayner, Y.Natanzon, Y.Kauffmann, Y.Amouyal .ACS Appl. Mater. Interfaces (2022)
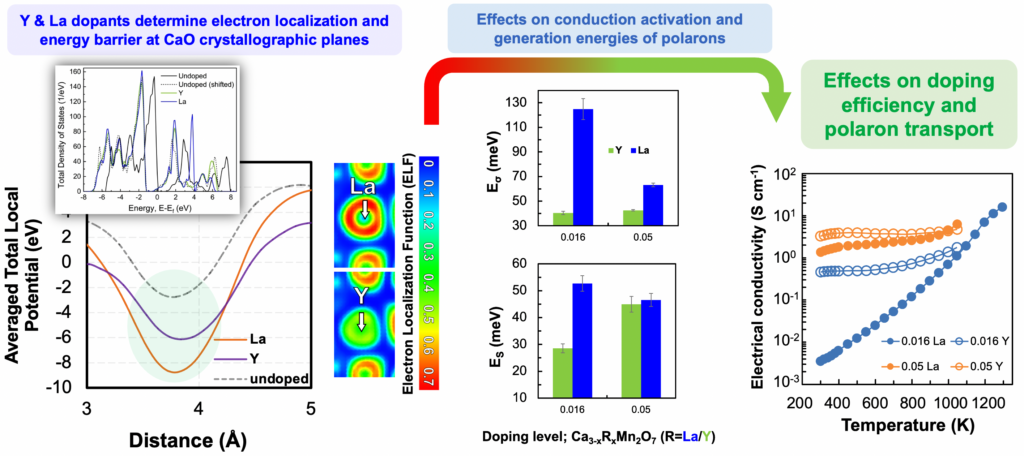
We investigate phase stability, microstructure, and thermoelectric transport of polycrystalline bulk Ca3−xRxMn2O7 samples prepared by standard solid-state reaction, where R = Y or La and 0 ≤ x ≤ 0.33. Ab-initio calculations predict that Y-doping at Ca-sites should reduce the potential energy barrier for electron transport, as opposed to La-doping. We find that Y-doping prompts transformation from Ca3Mn2O7 to Ca2MnO4, whereas La-doping is accompanied by no phase transformation. La-doping significantly hinders grain growth, for example, the average grain size decreases from 4.44 ± 0.24 to 1.20 ± 0.03 μm for x = 0 (undoped) and x = 0.33 upon La-doping, respectively. A. Azulay, T. Leibovitz, Y. Natanzon, Or Zabari, Y. Amouyal. J. Am. Ceram. Soc. (2022)
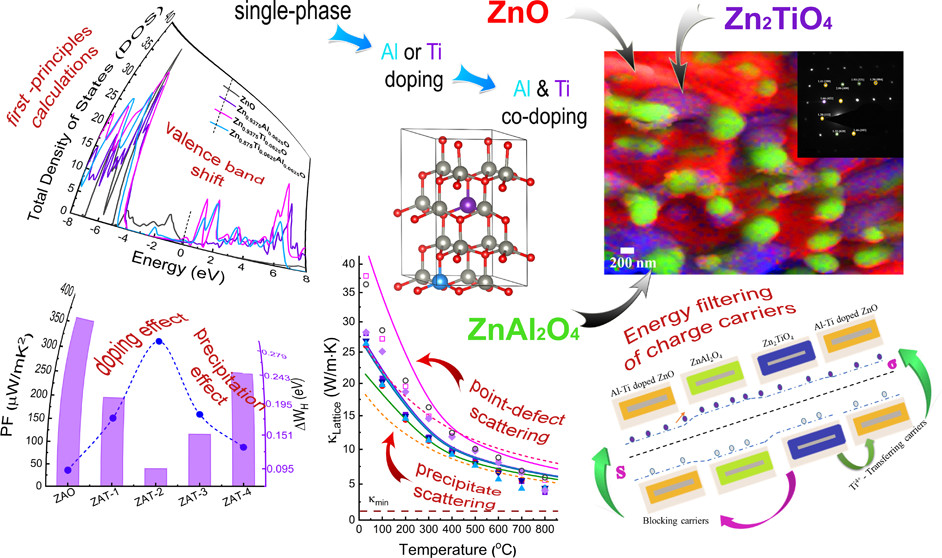
We observed unusual behavior, in which the Seebeck coefficient (S ) and electrical conductivity (σ) values increased simultaneously with temperature for Zn1–x–yAlxTiyO, originating from energy filtering of charge carriers due to both co-doping and the peculiar multiphase structure. High σ values were associated with the increase of electron concentration, while high S values were due to Fermi energy tuning and heavier effective masses initiated by Al and Ti co-doping. Besides increasing the PF, the multiphase structure played an essential role in reducing lattice thermal conductivity due to phonon scattering by the Umklapp, point defect, and second-phase mechanisms. Our approach yielded an increase of the TE figure of merit upon formation of a three-phase 2 wt % Ti-doped Zn0.98Al0.02O compound of ca. 10 times compared to the corresponding value attained for its two-phase ZnAl0.02O counterpart. C. Gayner, Y. Natanzon, Y. Amouyal. ACS Appl. Mater. Interfaces (2022)
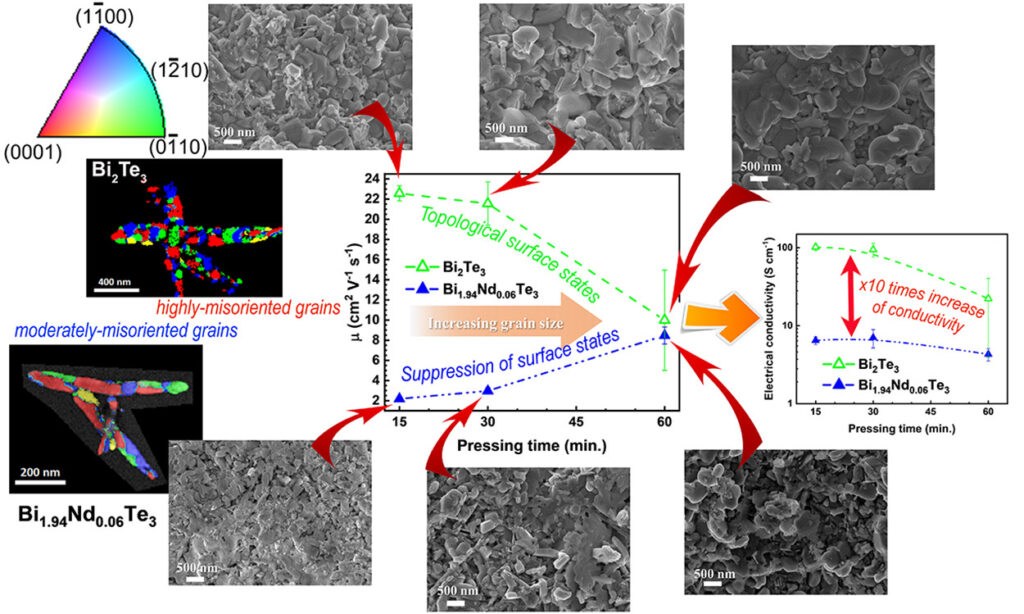
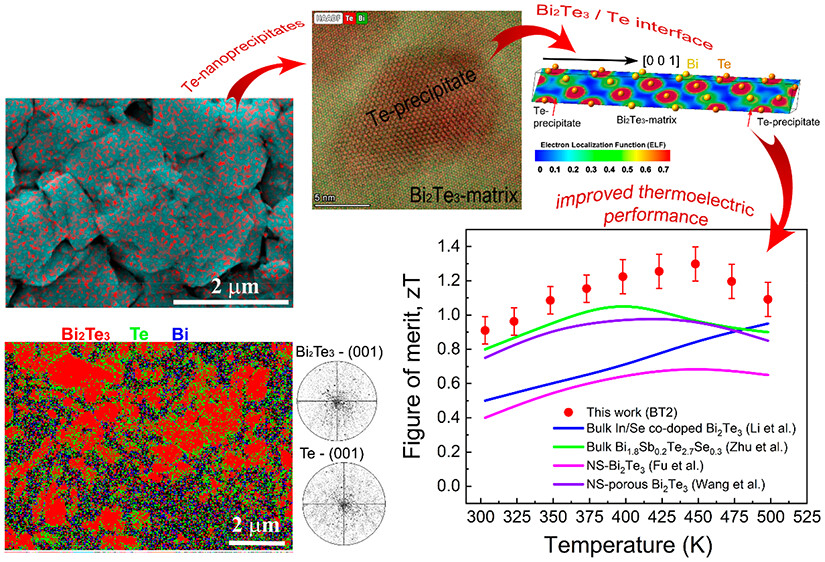
We found that the room-temperature values of electrical conductivity, Seebeck coefficient, and charge carrier mobility of the consolidated Bi2Te3 samples increase with the reduction of the average grain size. This phenomenon was observed earlier for Bi2Se3, which is a well-known TI material. We also found that Nd-doping reduces the electrical conductivity, Seebeck coefficient, charge carrier mobility, and thermal conductivity near room temperature (310 K). Interestingly, Nd-doping reverses the dependence of mobility on grain size compared to undoped Bi2Te3, which is associated with its magnetic moment. G. Solomon, E. Song, C. Gayner, J. A. Martinez, Y. Amouyal. ACS Appl. Nano Mater. 4, 4419 (2021)
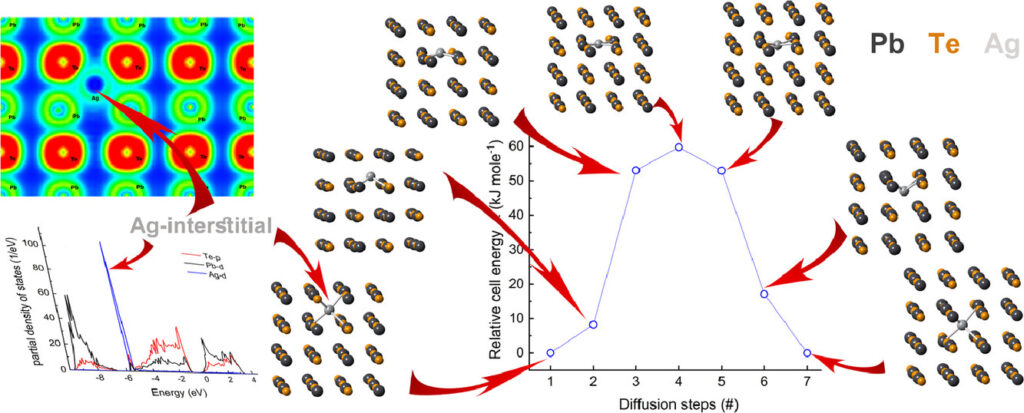
The diffusion coefficients of Ag in a PbTe matrix are evaluated utilizing first-principles calculations. Based on unique combination between calculations of point defect formation energies, electronic density of states (DOS), and electron localization function (ELF), we conclude that diffusion of Ag in PbTe in the interstitial mechanism is preferable compared to the vacancy or substitutional mechanism. We determine both the activation energy for diffusion and the pre-exponential diffusion coefficient for an interstitial mechanism, applying the transition state theory (TST), to be 1.08 × 10−5 cm2·s−1 and 52.9 kJ·mole−1, respectively. M. H. Dahan, A. Baranovskiy, Y. Natanzon, Y. Amouyal. Acta Materialia 202, 243-254 (2020).
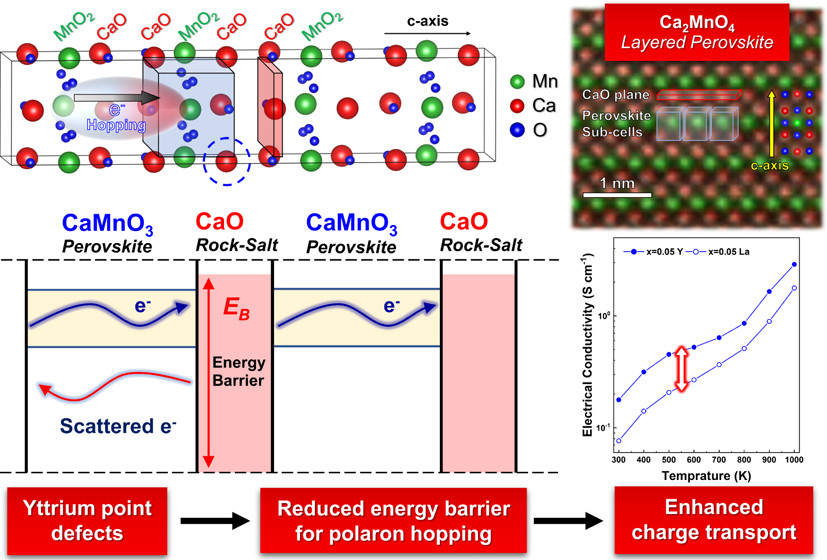
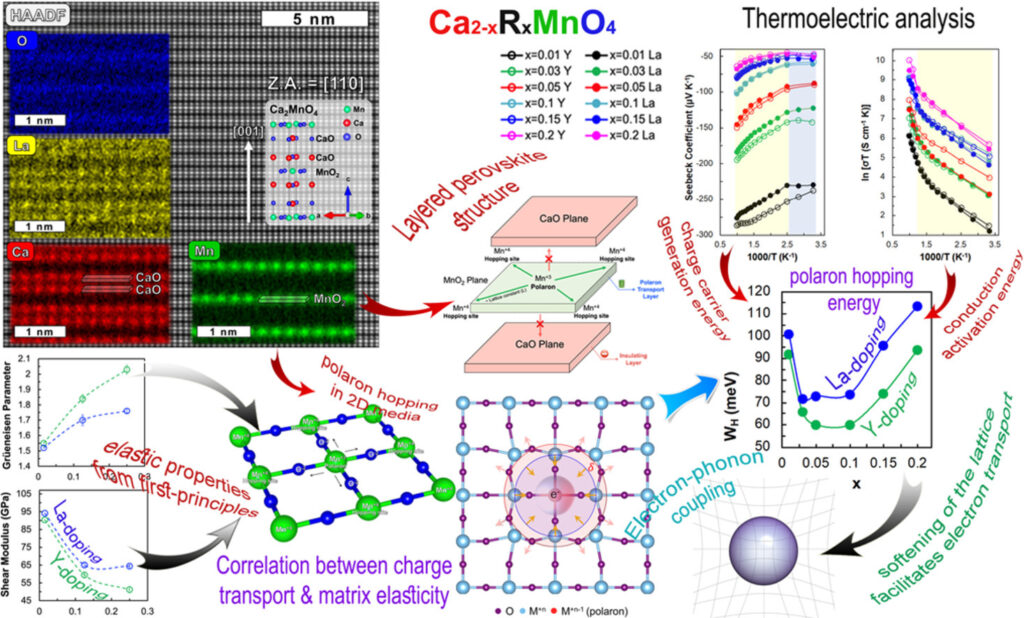
We apply the well-established phonon glass electron crystal (PGEC) concept in CaO(CaMnO3)m Ruddlesden–Popper layered perovskites by doping them with yttrium of lanthanum. This enables us to increase the transparency of CaO planes to electron transport at the same time while preserving their opacity to phonon transport. First-principles calculations indicate that the total local potential at CaO planes, where Y substitutes for Ca, is lower by ca. 50% compared to La substitution. Measurements of the electrical conductivity and Seebeck coefficients for Ca2–xRxMnO4 (R = La or Y; x = 0.01, 0.05, 0.1, and 0.15) bulk materials in the range of 300–1000 K confirm that compounds doped with Y exhibit higher electrical conductivity values than their La-doped counterparts. We attribute this to lower polaron hopping energy values (up to 23%) evaluated using the small polaron hopping model. A. Azulay, M. Wahabi, Y. Natanzon, Y. Amouyal. ACS Appl. Mater. Interfaces 12, 49768–49776 (2020).
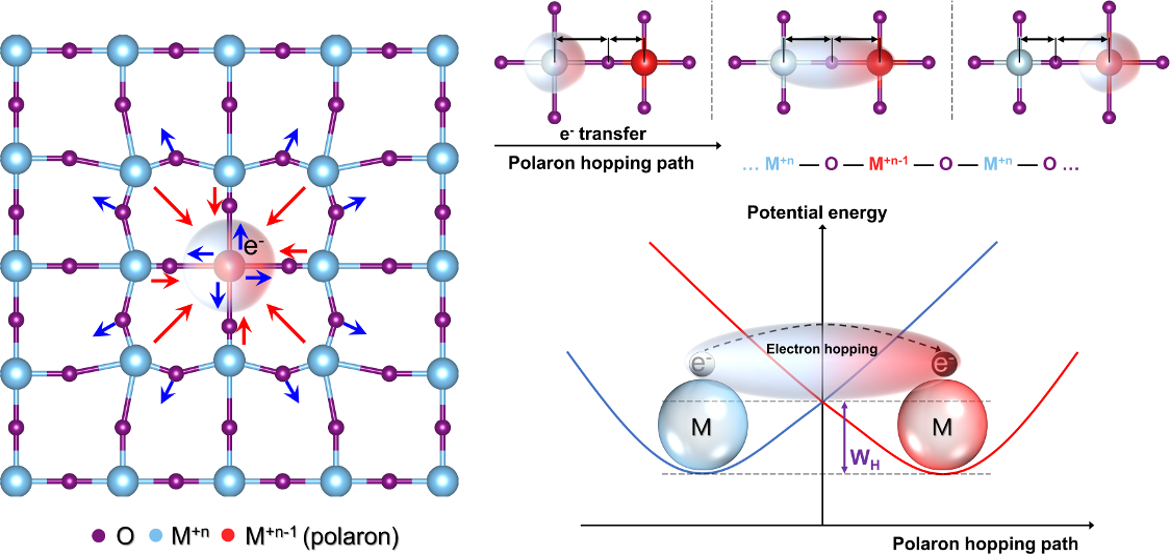 We review recent selected studies applying the density functional theory (DFT), and describe the merits and challenges in applying DFT for such calculations, highlighting the need to address both electronic and vibrational aspects. The vibrational component of the polaron is evaluated based on structural and total energy calculations, whereas the electronic component is derived from both total energy and electron density calculations. To address the most compelling challenge of calculating polaron properties using DFT, which is the issue of electron localization, we propose to employ calculations of selected vibrational properties, such as the sound velocity, shear modulus, and Grüneisen parameter, to represent the polaron hopping energy; all of which originate from the stiffness of inter-atomic bonds. Such methodology is expected to be more straightforward than the existing ones, however, demands standardization. Y. Natanzon, A. Azulay, Y. Amouyal. Isr. J. Chem. 60, 768 (2020).
We review recent selected studies applying the density functional theory (DFT), and describe the merits and challenges in applying DFT for such calculations, highlighting the need to address both electronic and vibrational aspects. The vibrational component of the polaron is evaluated based on structural and total energy calculations, whereas the electronic component is derived from both total energy and electron density calculations. To address the most compelling challenge of calculating polaron properties using DFT, which is the issue of electron localization, we propose to employ calculations of selected vibrational properties, such as the sound velocity, shear modulus, and Grüneisen parameter, to represent the polaron hopping energy; all of which originate from the stiffness of inter-atomic bonds. Such methodology is expected to be more straightforward than the existing ones, however, demands standardization. Y. Natanzon, A. Azulay, Y. Amouyal. Isr. J. Chem. 60, 768 (2020).
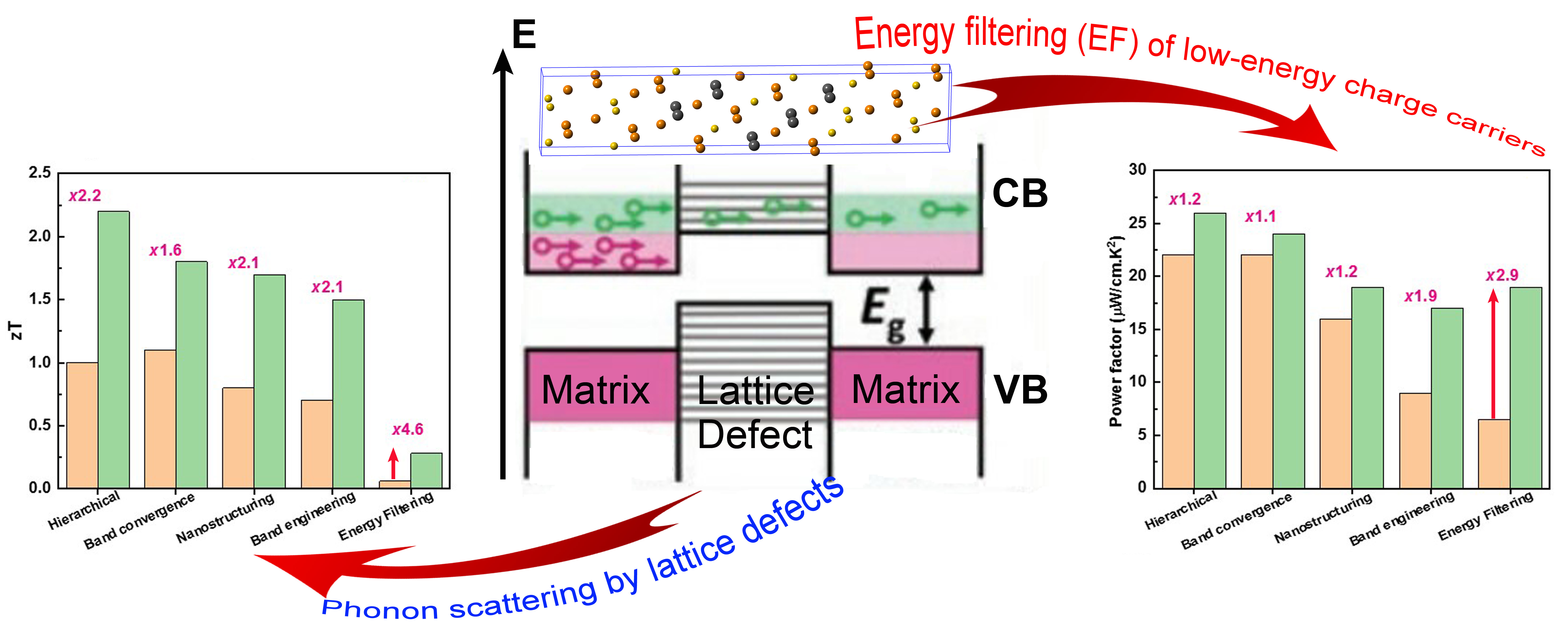
Various concepts and paradigms regarding energy filtering effect are reviewed. Ways to optimize thermoelectric transport via energy filtering are also discussed. The authors demonstrate the potential of energy filtering for thermoelectric energy conversion. Although this effect theoretically demonstrates improvement of the thermopower, applying it poses certain constraints, which demands further research. Predicated on data documented in literature, the unusual dependence of the thermopower and conductivity upon charge carrier concentrations can be altered through the energy filtering approach. Upon surmounting the physical constraints discussed in this article, thermoelectric materials research may gain a new direction to enhance the power factor and thermoelectric figure of merit. C. Gayner, Y. Amouyal. Adv. Func. Mater. 29, 1901789 (2019)
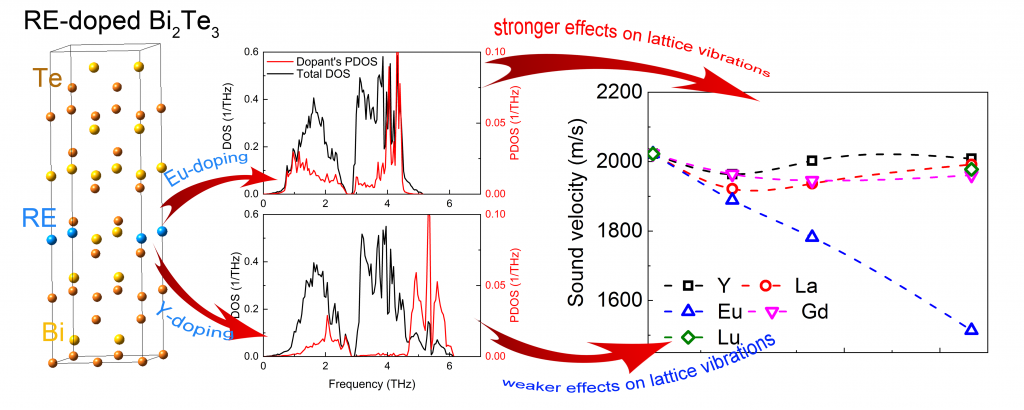
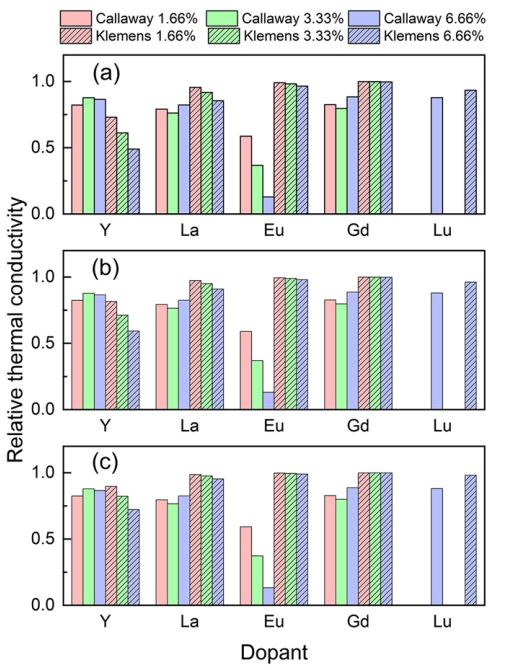
The density functional theory is a powerful tool for thermal conductivity evaluation. This procedure requires consideration of both Grüneisen parameter and sound velocity. Doping of bismuth telluride with rare earth elements reduce thermal conductivity. Impurity phonon modes appear at frequencies up to 2 THz. The divalent impurities exhibit stronger effects compared to trivalent ones. A. Baranovskiy et al., Advanced Theory and Simulations 1 (12), 1800162 (2019).
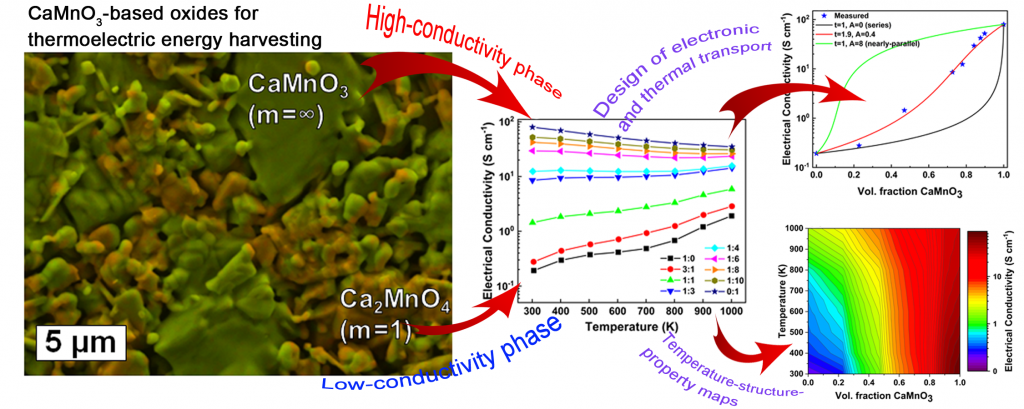
Oxide thermoelectric materials are promising for waste heat recovery at elevated temperatures. Motivated by the continuing efforts seeking for high-efficiency n-type oxides, we study Nb-doped n-type CaMnO3 (m = ∞) and Ca2MnO4 (m = 1), which are derivatives of the Ruddlesden-Popper CaO(CaMnO3)m-series featuring extremely opposite transport coefficients. We synthesize composite materials based on these two phases to correlate between the thermoelectric properties, microstructure evolution, and composition of the material, and determine the optimum sintering temperature. Then, we measure both thermal and electronic transport coefficients, and perform a thorough general effective medium (GEM) analysis. Interestingly, we find that most ratios obey to the GEM behavior, where deviations are elucidated in terms of interfacial effects. This study provides us with tools for identifying the significance of bulk vs. interfacial effects in design of composite materials with controllable transport properties. A. Azulay and Y. Amouyal. Acta Mater. 164 481-492 (2019)
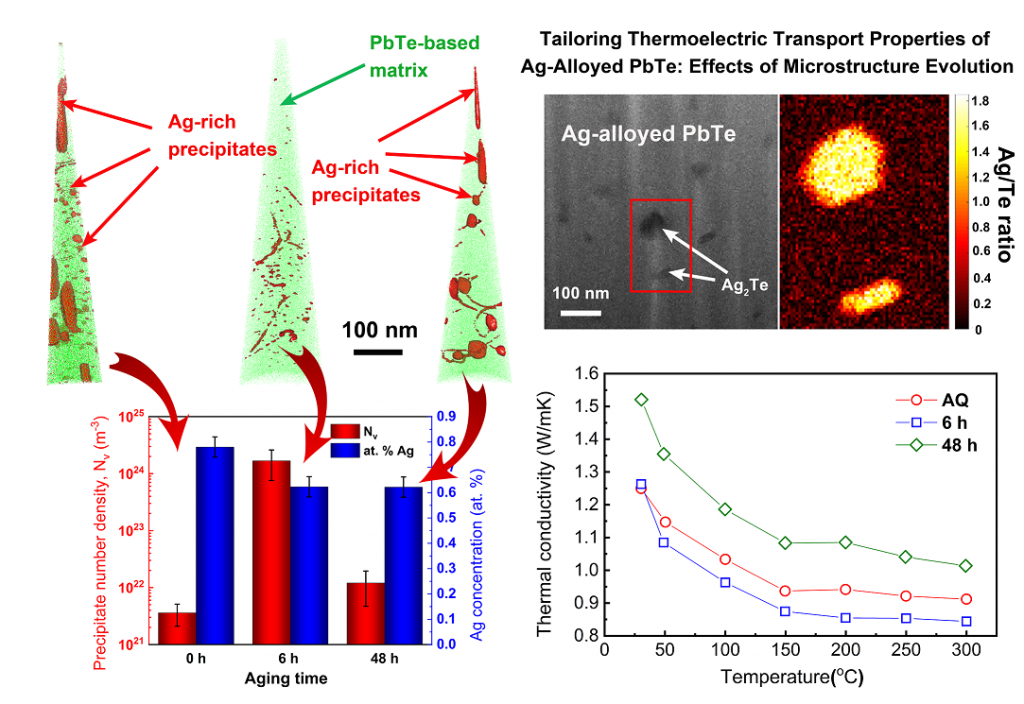
Complementary scanning transmission electron microscopy (S/TEM) and atom probe tomography (APT) analysis reveal nanometer-size Ag-rich precipitates in PbTe-based thermoelectric matrix of high number density. The thermal and electrical conductivities, as well as the Seebeck coefficients, are found to be highly sensitive to the microstructure and its temporal evolution, e.g., the number density of Ag-rich precipitates increases by ca. 3 orders of magnitude and reaches (1.68 ± 0.92) × 1024 m−3 upon aging at 380 °C for 6 h, which is pronounced by reduction in thermal conductivity to a value as low as 0.85 W m−1 K−1 at 300 °C. This guides predictive tools for further design of materials for energy harvesting. A. Sheskin et al. ACS Appl. Mater. Inter., 10 (45) 38994-39001 (2018). DOI: 10.1021/acsami.8b15204
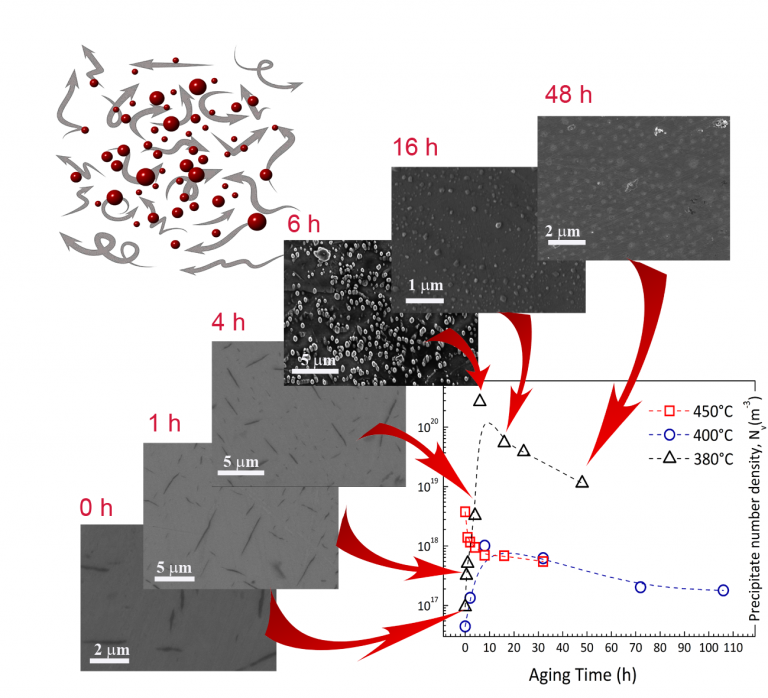
A commonly used approach to enhance thermoelectric efficiency adapted from physical metallurgy is formation of highly-dispersed precipitates in the matrix for phonon scattering. Selection of the appropriate alloy composition, as well as heat treatment temperature and duration, enables delicate control of the microstructure evolution kinetics, thereby achieving minimum lattice thermal conductivity. More details: Y. Amouyal et al., Progress Report: Physical Metallurgy Inspired Nano-Features for Enhancement of Thermoelectric Conversion Efficiency, Advanced Theory and Simulations July 2018, 1800072. DOI: 10.1002/adts.201800072
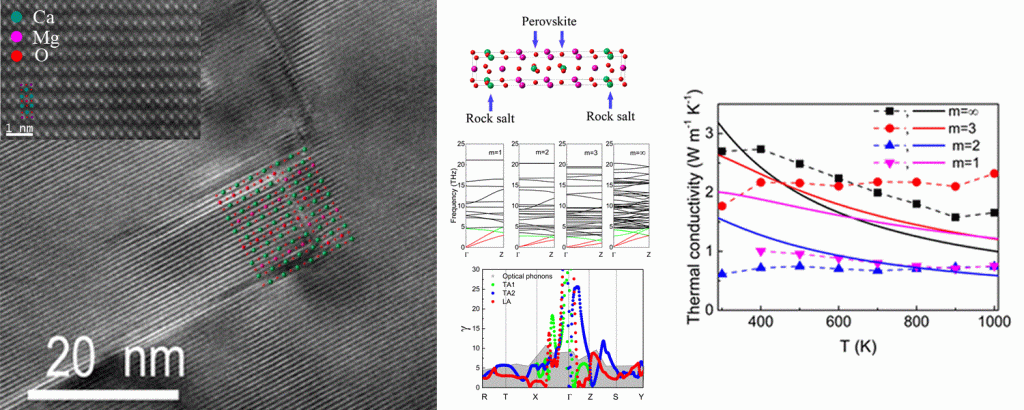
Vibrational properties of CaO(CaMnO3)m (m = 1, 2, 3, and ∞) thermoelectric (TE) oxides for high-temperature energy conversion applications are studied both experimentally and computationally. It is found that thermal conductivity of CaO(CaMnO3)m compounds is governed by phonon scattering on CaO/CaMnO3 boundaries for m = 1, 2, and 3 and by Umklapp processes for m = ∞. Thermal transport in this system is strongly dominated by acoustic phonon modes possessing much larger Grüneisen parameters (γ) compared to optical ones. A. Baranovskiy et al. Nano Energy 47 451-462 (2018). DOI: 10.1016/j.nanoen.2018.02.054
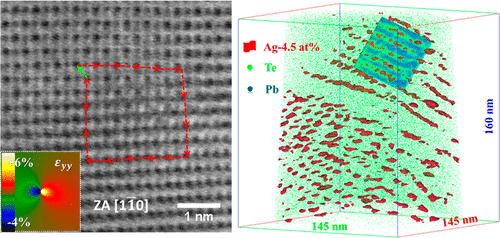 Here, we correlate transmission Kikuchi diffraction and (scanning) transmission electron microscopy in conjunction with atom probe tomography to investigate the local structure and chemical composition of dislocations in a thermoelectric Ag-doped PbTe compound. Our investigations indicate that Ag atoms segregate to dislocations with a 10-fold excess of Ag compared with its average concentration in the matrix. Thermal conductivity is evaluated applying laser flash analysis, and is correlated with theoretical calculations based on the Debye–Callaway model, demonstrating that these Ag-decorated dislocations yield stronger phonon scatterings. These findings reduce the knowledge gap regarding the composition of dislocations needed for theoretical calculations of phonon scattering and pave the way for extending the concept of defect engineering to thermoelectric materials. Y. Yu et al. ACS Applied Materials & Interfaces, 10 (4), 3609 (2018).
Here, we correlate transmission Kikuchi diffraction and (scanning) transmission electron microscopy in conjunction with atom probe tomography to investigate the local structure and chemical composition of dislocations in a thermoelectric Ag-doped PbTe compound. Our investigations indicate that Ag atoms segregate to dislocations with a 10-fold excess of Ag compared with its average concentration in the matrix. Thermal conductivity is evaluated applying laser flash analysis, and is correlated with theoretical calculations based on the Debye–Callaway model, demonstrating that these Ag-decorated dislocations yield stronger phonon scatterings. These findings reduce the knowledge gap regarding the composition of dislocations needed for theoretical calculations of phonon scattering and pave the way for extending the concept of defect engineering to thermoelectric materials. Y. Yu et al. ACS Applied Materials & Interfaces, 10 (4), 3609 (2018).
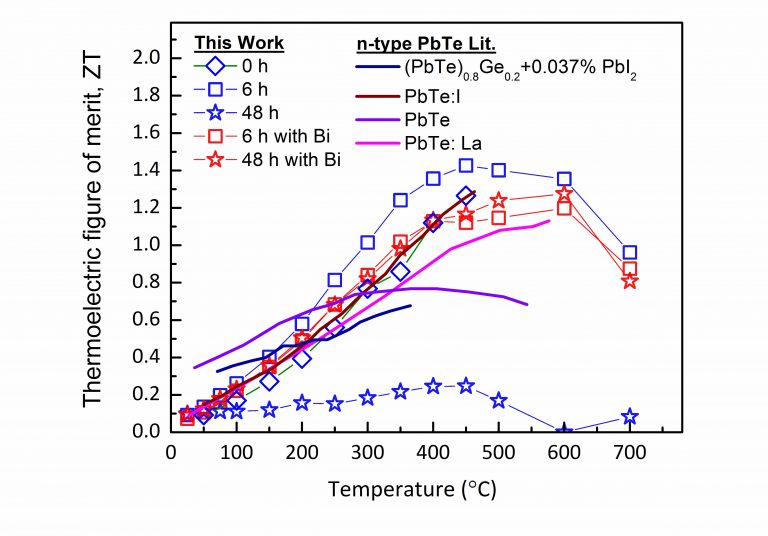
Significant enhancement of ZT for n-type PbTe-based compound due to controlled nucleation and growth of Ag-rich precipitates. T. Grossfeld, A. Sheskin, Y. Gelbstein, and Y. Amouyal, Crystals 7 (9), 281 (2017) http://www.mdpi.com/2073-4352/7/9/281
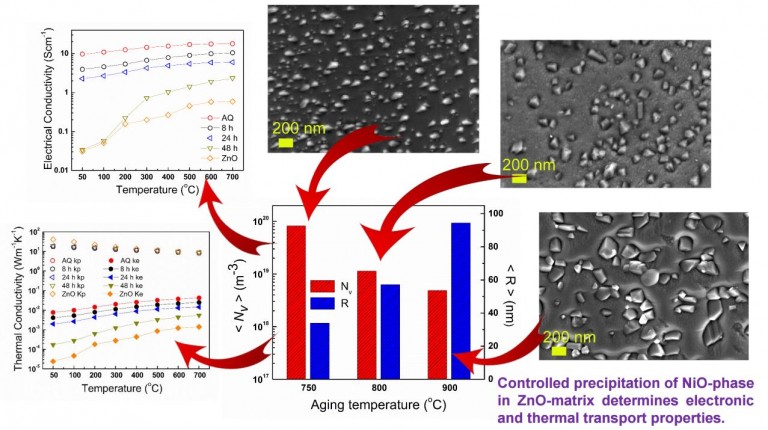
Effects of precipitation on the thermal conductivity of nickel-doped zinc oxide for thermoelectric applications. I. Koresh and Y. Amouyal, J. Eur. Ceram. Soc. 37 (11), 3541-3550 (2017)
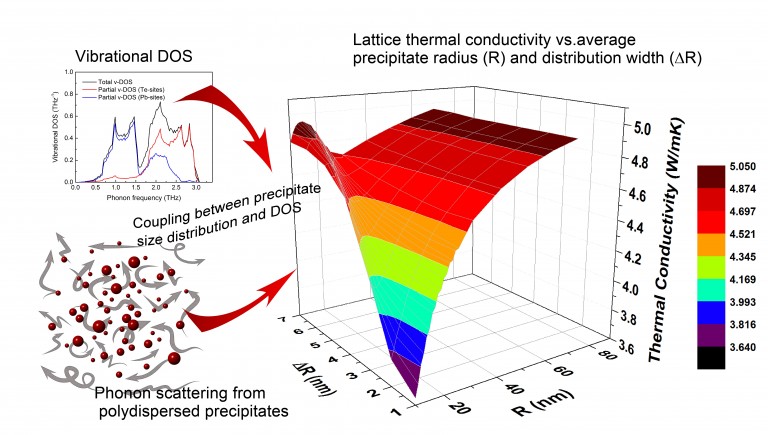
A Practical Approach to Evaluate Lattice Thermal Conductivity in Two-Phase Thermoelectric Alloys for Energy Applications. Y. Amouyal, Materials 10 (4), 386 (2017).
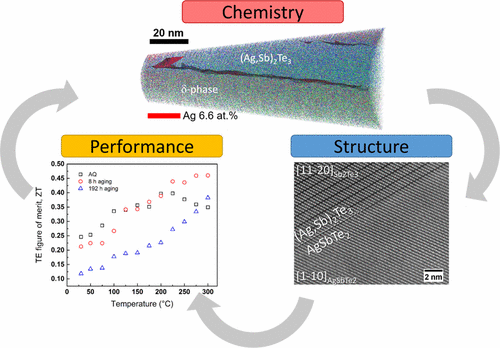
Role of nanostructuring and microstructuring in silver antimony telluride compounds for thermoelectric applications. O. Cojocaru-Mirédin, L. Abdellaoui, M. Nagli, S. Zhang, Y. Yu, C. Scheu, D. Raabe, M. Wuttig, and Y. Amouyal, ACS Applied Materials & Interfaces 9, 14779-14790 (2017).
The longitudinal acoustic
Optical
The longitudinal acoustic (top) and optical (second) modes of lattice vibrations in cubic Pm-3m CaMnO3, simulated from density functional theory (DFT) calculations. The Ca, Mn, and O atoms are indicated by blue, aqua, and red spheres, respectively. It is observed that Ca-atoms mainly contribute to the acoustic modes, whereas the optical ones are dominated by oxygen.
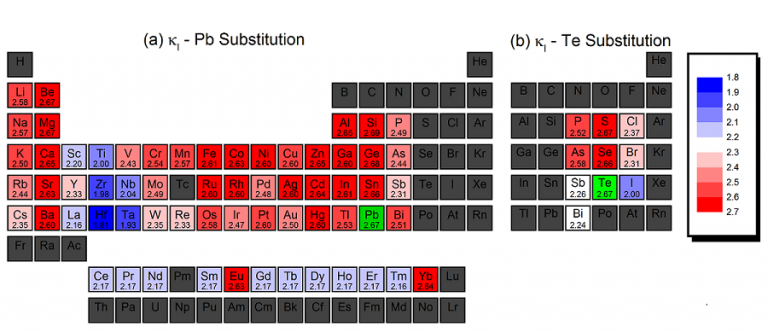
A color-coded periodic table representing the lattice thermal conductivity calculated for 1.6 at. % X-doping at 700K for (a) Pb-site substitutions, XPb31Te32 and (b) Te-site substitutions, XPb32Te31. The values in the diagram are expressed in Wm-1K-1 , and are calculated based on an average phonon relaxation time of 10-11 s. E. Joseph and Y. Amouyal, J. Appl. Phys. 117 (17), 175102 (2015).
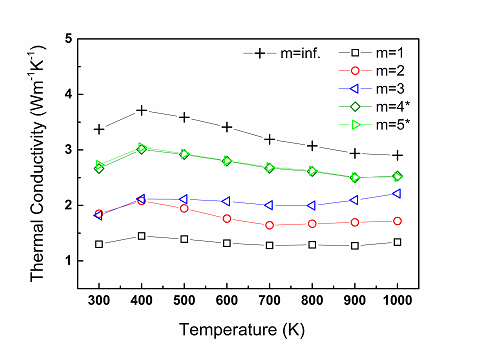
The temperature-dependent thermal conductivity measured for the Nb-doped compounds of different periodicities, having the structural form (CaMnO3)mCaO ,using the laser flash analysis (LFA) technique: m=∞ (plus markers), m=1 (black), m=2 (red), m=3 (blue), m=4* (dark green), and m=5* (light green). A. Graff and Y. Amouyal, Appl. Phys. Lett. 105(18), 181906 (2014).
The calculated temperature dependent (a) lattice thermal conductivity based on the calculated sound velocity, and (b) total lattice thermal conductivity for La-doped PbTe of different concentrations. E. Joseph and Y. Amouyal, J. Elect. Mater. 44 (6), 1460 (2015).
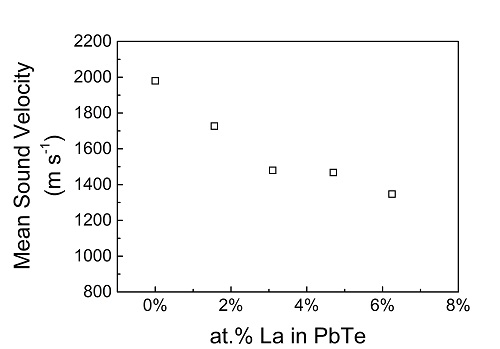
The sound velocity averaged over the different modes calculated for a La-doped PbTe lattice, as a function of the doping level. A significant decrease of the sound velocity with increasing doping level is observed. E. Joseph and Y. Amouyal, J. Elect. Mater. 44 (6), 1460 (2015).
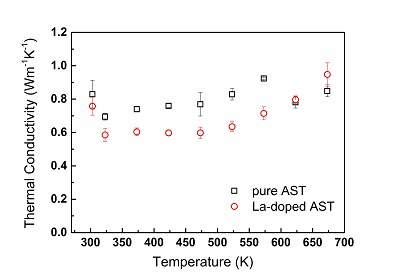
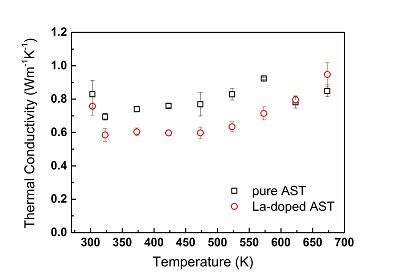
Temperature-dependent thermal conductivity, as measured by use of the LFA method, for a ternary alloy AgSbTe2-based alloy (AST, squares) and a quaternary, La-doped, alloy (La-doped AST, circles). A notable difference between the thermal conductivity values is apparent along a wide temperature range Y. Amouyal, J. Elect. Mater. 43 (10), 3772 (2014).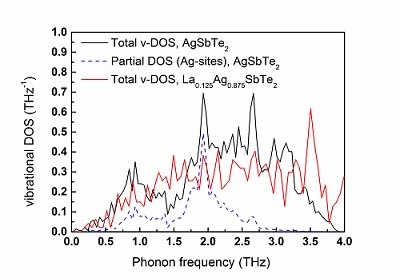
The vibrational density of states (v-DOS) calculated, from first-principles, for the AgSbTe2 phase. The total v-DOS appears in black and the partial v-DOS associated with the Ag-sublattice vibrations is represented by the blue dashed curve. The noticeable overlap at approximately 2 THz implies that the primary vibrational peak in the AgSbTe2 v-DOS curve originates from the Ag sublattice. Attenuation of the v-DOS as a result of La-doping is observed for a La0.125Ag0.875SbTe2 lattice, denoted by the red line. Y. Amouyal, J. Elect. Mater. 43 (10), 3772 (2014).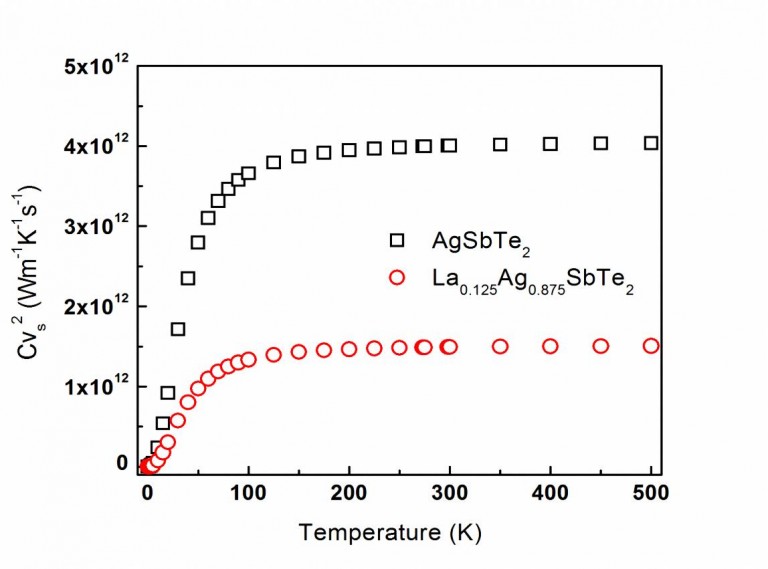
The temperature-dependence of the Cv·vs2-products (Cv: heat capacity; vs: sound velocity) calculated for the AgSbTe2– and La0.125Ag0.875SbTe2-compounds from first-principles, marked with empty squares and circles, respectively. This product is a significant factor determining thermal conductivity; the marked difference between both plots evidently indicates the effects of La-doping on lowering thermal conductivity. Y. Amouyal, Computational Materials Science 78 (2013) 98.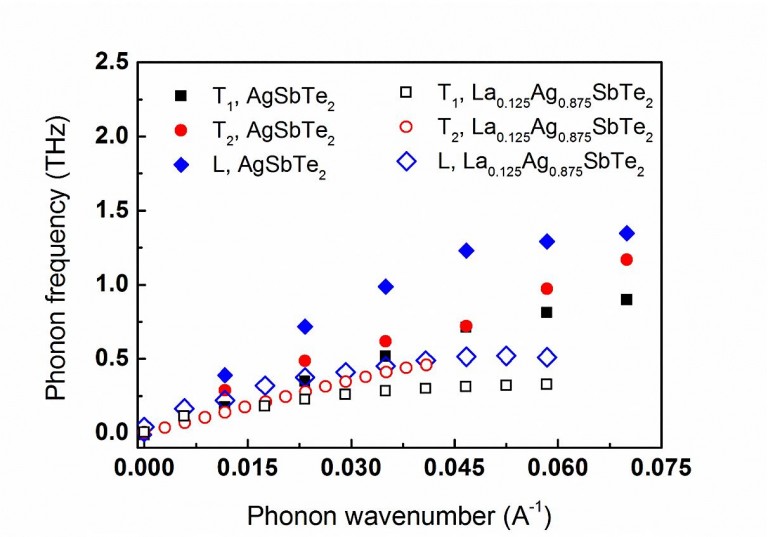
The dispersion ω(k) – curves of the acoustic phonons calculated for both AgSbTe2 – and La0.125Ag0.875SbTe2– compounds close to the Γ-point. It is indicated that the slopes of the one longitudinal (filled diamonds) and two transverse (filled circles and squares) acoustic modes of the AgSbTe2-lattice are greater than those of the La0.125Ag0.875SbTe2-lattice (empty diamonds, circles and squares, respectively). Y. Amouyal, Computational Materials Science 78 (2013) 98.
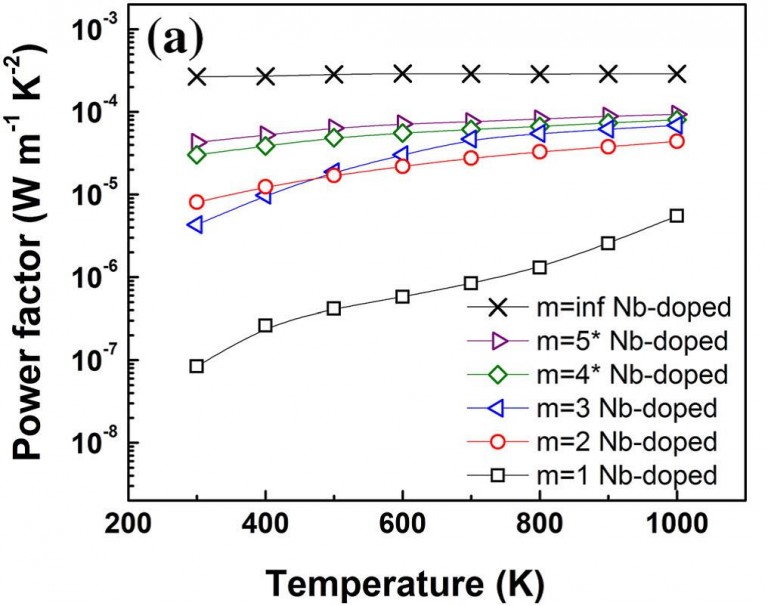
Temperature-dependent power factor (PF) values calculated for Nb-doped CaO(CaMnO3)m compounds with different periodicities of m = ∞ (inf., cross markers), m = 1 (black squares), m = 2 (red circles), m = 3 (blue left-triangles), m = 4* (green diamonds), and m = 5* (purple right-triangles). A. Graff & Y. Amouyal J. Electron. Mater. 45 (3), 1508 (2016).
The longitudinal acoustic
Optical
La0.125Ag0.875SbTe2
AgSbTe2
The longitudinal acoustic modes of lattice vibrations in pure and lanthanum-doped 2x2x2 AgSbTe2-based supercells, simulated from density functional theory (DFT) calculations. The La, Ag, Sb, and Te atoms are indicated by aqua, gray, yellow, and orange spheres, respectively. It is observed that La-substitution defects suppress lattice vibrations, thereby reducing lattice thermal conductivity. Y. Amouyal, Computational Materials Science 78 (2013) 98.

A high-resolution scanning electron microscope (SEM) micrograph of oriented Sb2Te3-precipitates in an Ag-Sb-Te-based matrix aged at 400 °C for 0.5 h.

A high-resolution scanning electron microscope (SEM) micrograph of a 4 at% Nb -doped CaMnO3-based Ruddlesden-Popper compound produced by solid state reaction of powders and sintering at 1400 °C. A. Graff and Y. Amouyal, Applied Physics Letters 105 (18), 181906 (2014).